Our science
Cortical Malformations and Focal Epilepsies
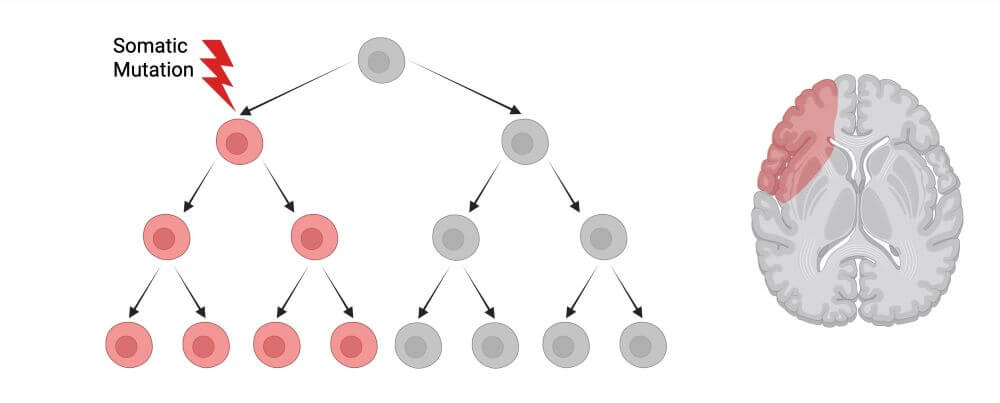
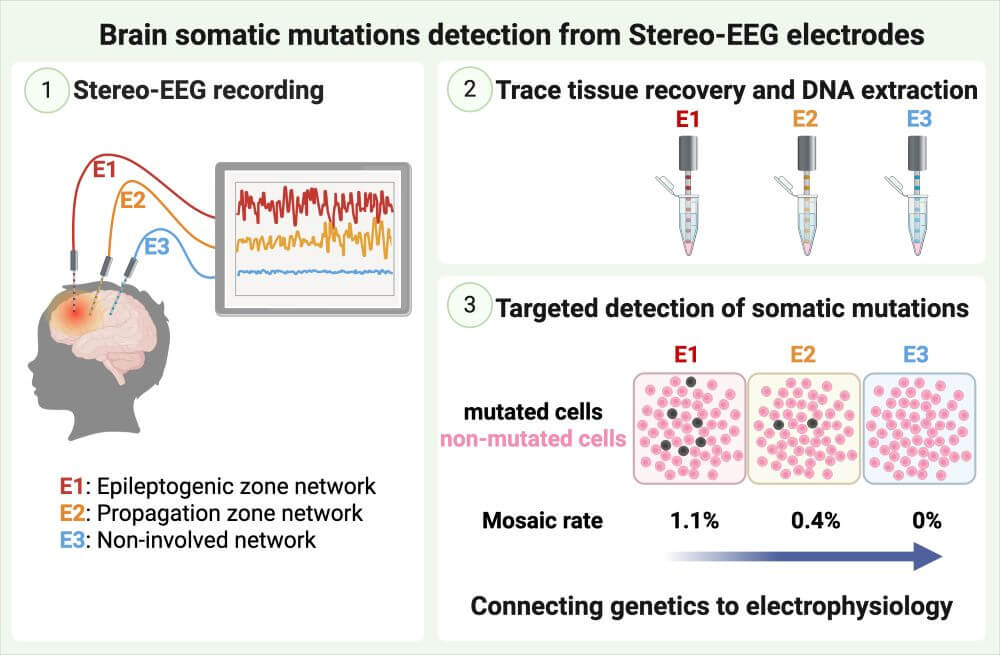
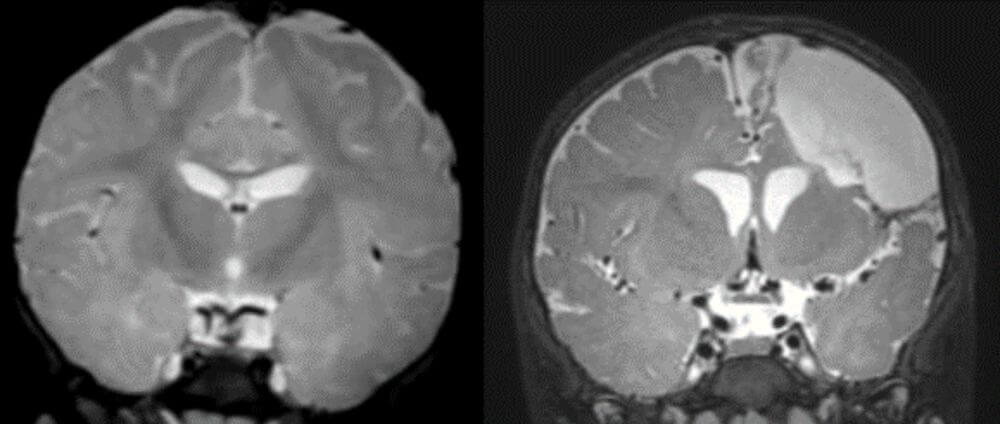
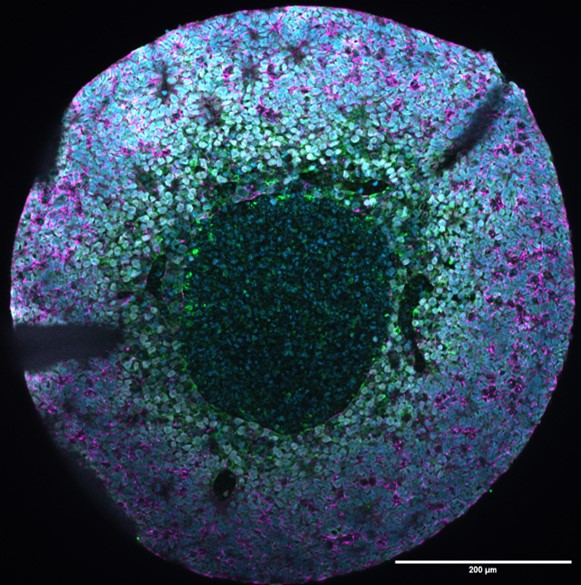
Unraveling the genetic etiology of cortical malformations and epilepsies
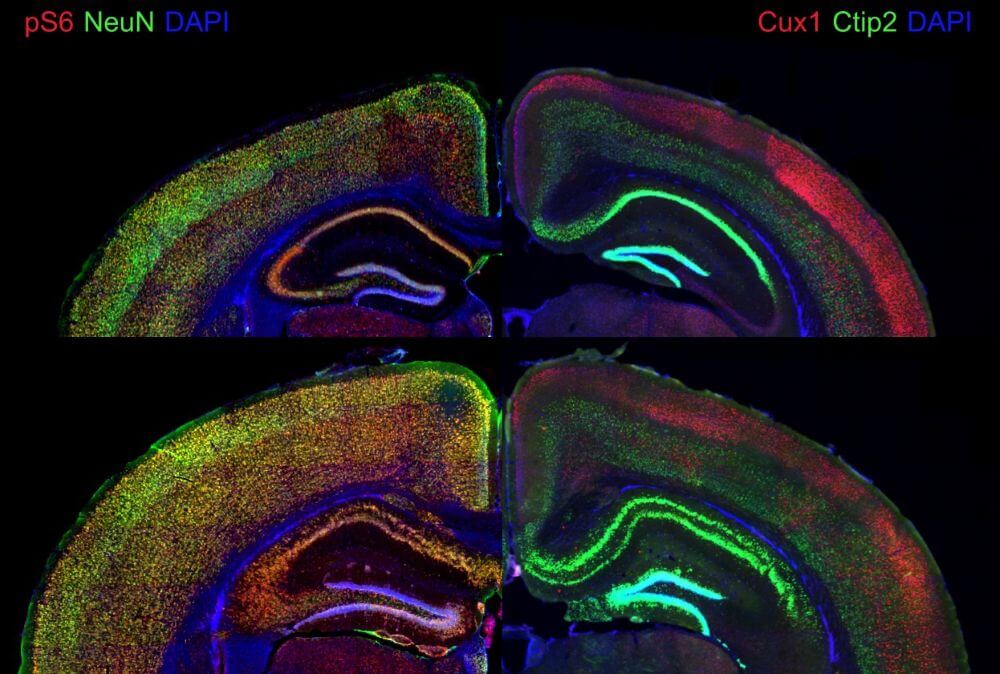
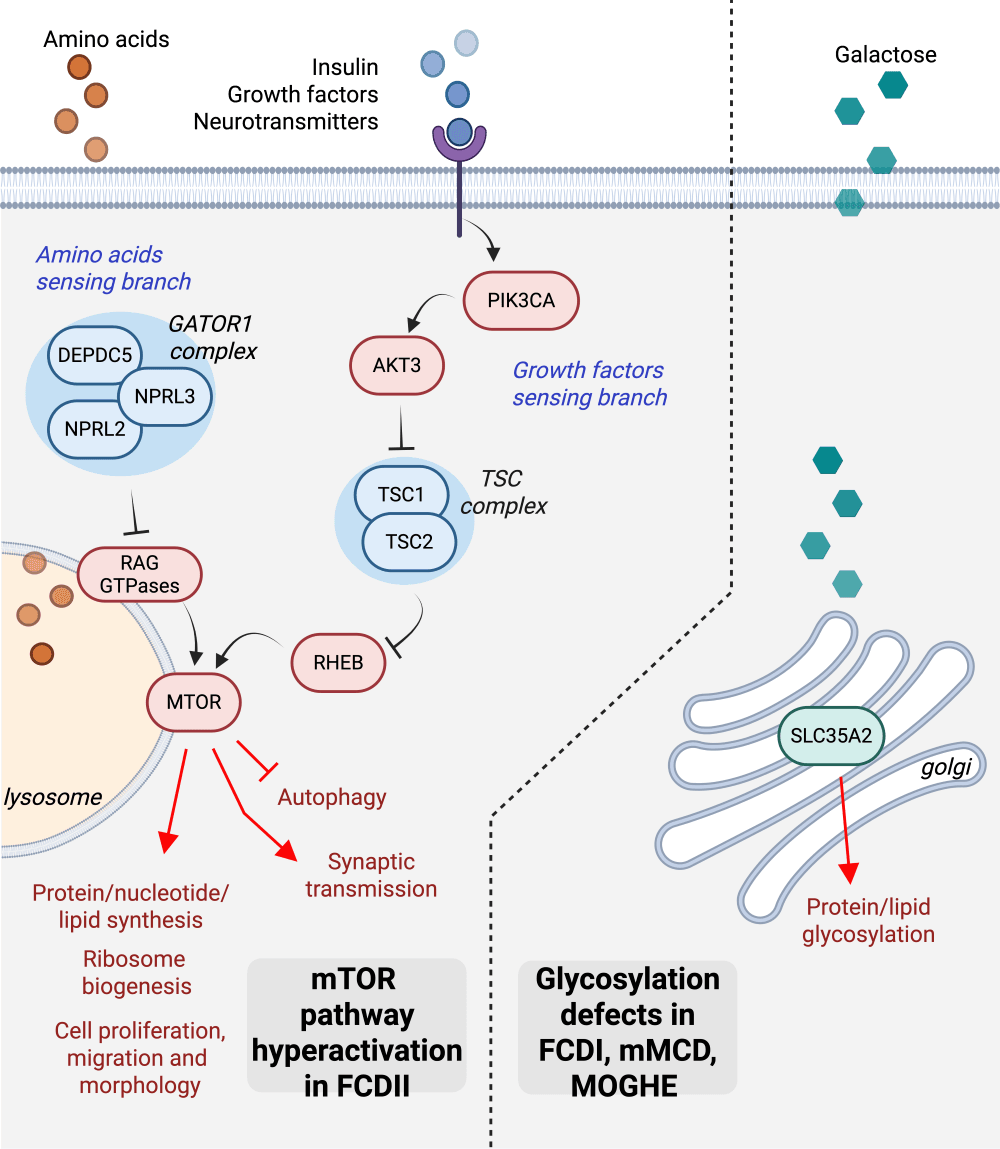
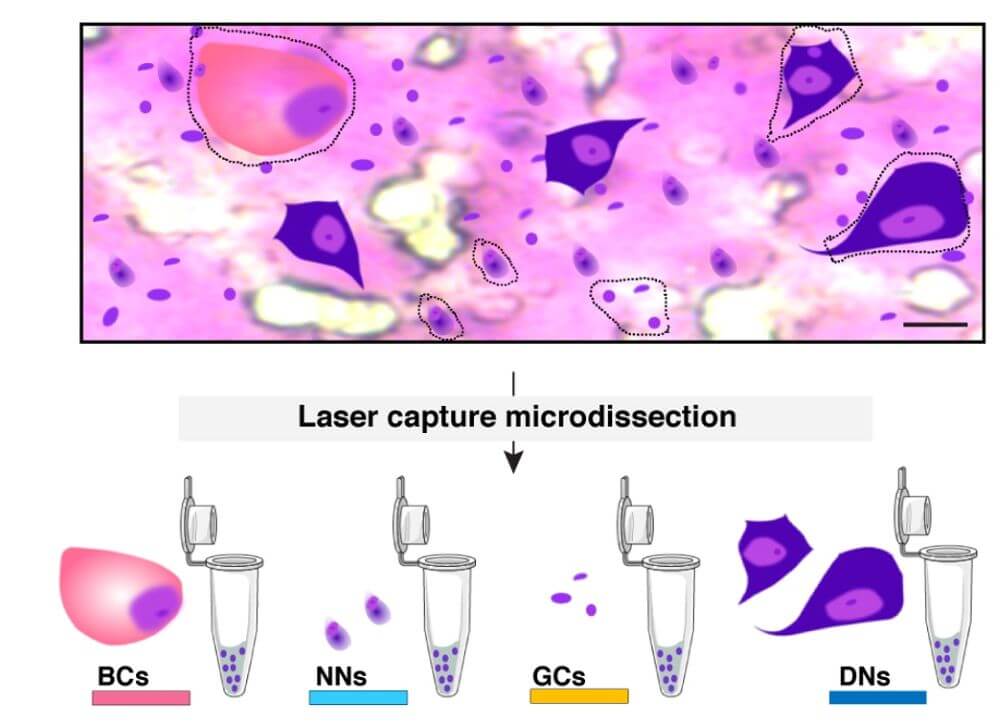
Brain mosaic mutations in mTOR pathway components cause FCD type II (FCDII), characterized by dysmorphic neurons and balloon cells exhibiting mTOR hyperactivation. Through ultra-deep gene panel sequencing and whole exome/long-read whole genome sequencing, we identify FCDII-causing somatic mutations in resected brain tissues and SEEG electrodes adherent tissues. We combine innovative molecular approaches including laser microdissection, droplet digital PCR, and single-cell multi-omics to map mutation-carrying cell populations and their molecular signatures within the altered brain architecture.
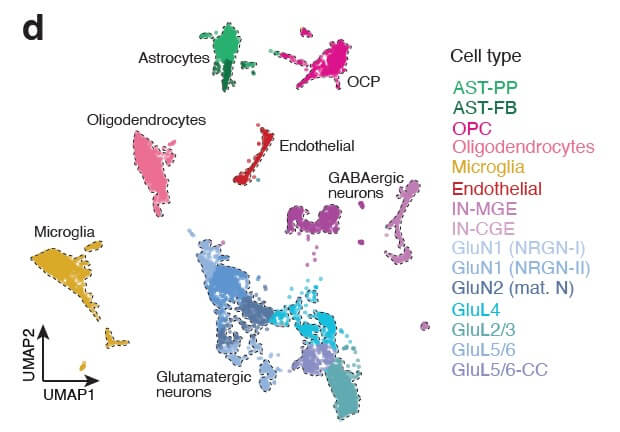
Our integrated approach encompasses genotyping and transcriptomics analyses at the single-cell level, enabling precise characterization of mutation burden and its functional consequences across individual cells within dysplastic tissue. To understand how genetic alterations translate into aberrant network activity, we employ multi-electrode array (MEA) recordings in acute cortical slices from FCDII patients, complemented by video-EEG monitoring for in vivo seizure characterization.
We are further employing proteomics and glycomics methodologies to investigate MOGHE pathophysiology, as SLC35A2 mutations impair UDP-galactose transport and compromise N-linked glycosylation processes. Through mass spectrometry-based analytical techniques, we characterize aberrant protein glycosylation profiles and examine their consequent impacts on cellular functionality and intercellular signaling within MOGHE tissues.
Our research aims to decode the genetic basis of these conditions and develop a comprehensive understanding of their molecular mechanisms, ultimately identifying potential therapeutic targets.
Modeling neurodevelopmental epilepsies with human cortical organoids
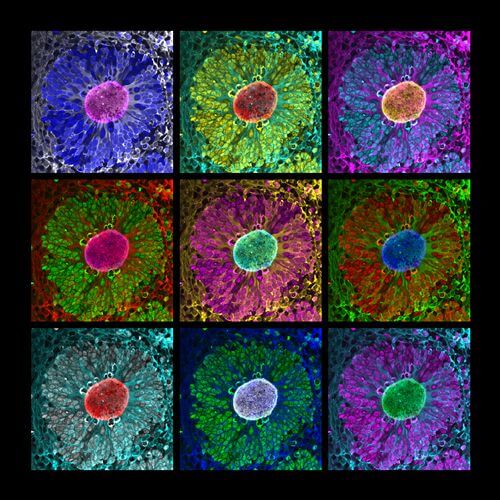
By combining cell reprogramming and genome engineering technologies, we generate iPSC-derived cortical organoids and assembloids to model focal and mosaic cortical malformations in vitro. We leverage these approaches to develop human neuronal models of FCDII caused by mTOR pathway mutations, and MOGHE caused by SLC35A2 mutations. Our research seeks to understand how mosaic pathogenic mutations alter cortical development and impact neuronal network activity through integrated single-cell and molecular analyses.
In vivo modeling of somatic mutations
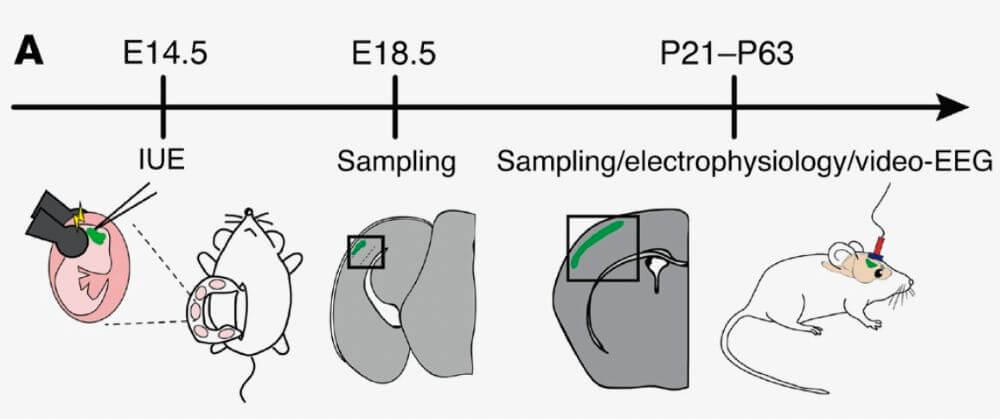
Mosaic hyperactivation of the mTOR signaling pathway leading to abnormal cells is at the heart of the pathogenic process in FCDII. We study pathogenic mechanisms using in utero electroporation (IUE) to generate genetic mouse models of cortical mosaic mTOR activation through either CRISPR-mediated knockout of Depdc5 (Depdc5fKO) or IUE of MTOR mutations, where only a subset of progenitors are mutated, allowing cellular mosaicism at the electroporation site. These complementary approaches enable us to model both loss-of-function and gain-of-function mechanisms within the mTOR signaling cascade. Our goal is to understand how mTOR activation leads to epileptogenesis through integrated functional, molecular, and behavioral analyses.